Our featured projects reflect the scientific endeavors of the Tau Consortium scientists. You can view our publications below and their respective datasets can be accessed through the AD Discovery Portal and analyzed using AD Workbench.
bioRxiv
CRISPR screens in iPSC-derived neurons reveal principles of tau proteostasis
Avi J. Samelson, Nabeela Ariqat, Justin McKetney, Gita Rohanitazangi, Celeste Parra Bravo, Rudra Bose, Kyle J. Travaglini, Victor L. Lam, Darrin Goodness, Gary Dixon, Emily Marzette, Julianne Jin, Ruilin Tian, Eric Tse, Romany Abskharon, Henry Pan, Emma C. Carroll, Rosalie E. Lawrence, Jason E. Gestwicki, David Eisenberg, Nicholas M. Kanaan, Daniel R. Southworth, John D. Gross, Li Gan, Danielle L. Swaney, Martin Kampmann
Abstract
Aggregation of the protein tau defines tauopathies, which include Alzheimer’s disease and frontotemporal dementia. Specific neuronal subtypes are selectively vulnerable to tau aggregation and subsequent dysfunction and death, but the underlying mechanisms are unknown. To systematically uncover the cellular factors controlling the accumulation of tau aggregates in human neurons, we conducted a genome-wide CRISPRi-based modifier screen in iPSC-derived neurons. The screen uncovered expected pathways, including autophagy, but also unexpected pathways, including UFMylation and GPI anchor synthesis. We discover that the E3 ubiquitin ligase CUL5SOCS4 is a potent modifier of tau levels in human neurons, ubiquitinates tau, and is a correlated with vulnerability to tauopathies in mouse and human. Disruption of mitochondrial function promotes proteasomal misprocessing of tau, which generates tau proteolytic fragments like those in disease and changes tau aggregation in vitro. These results reveal new principles of tau proteostasis in human neurons and pinpoint potential therapeutic targets for tauopathies.
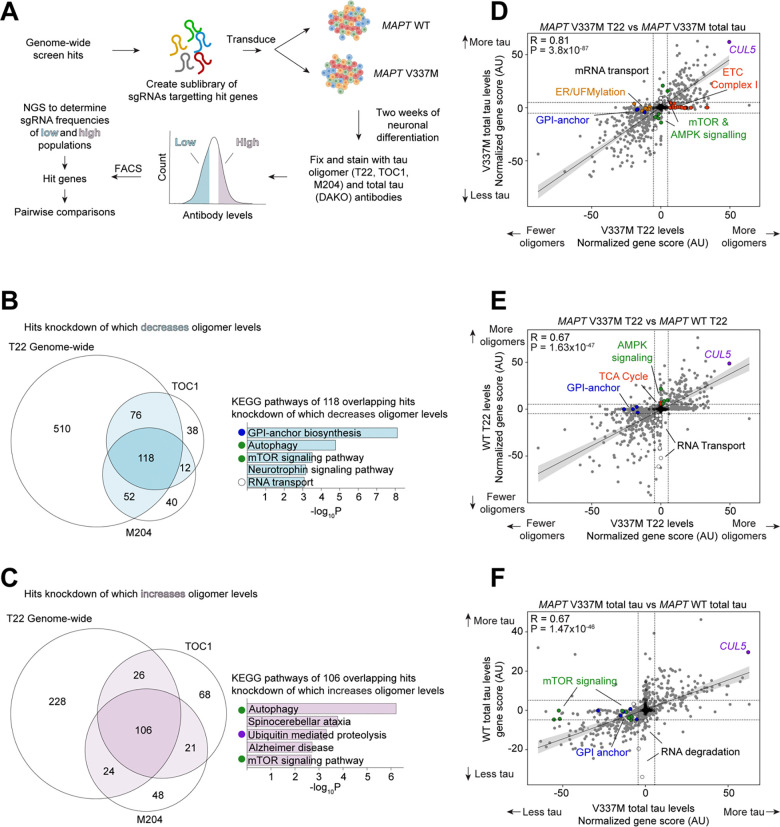
Neuron
Single-nucleus RNA sequencing demonstrates an autosomal dominant Alzheimer’s disease profile and possible mechanisms of disease protection
Maria Camila Almeida, Sarah J Eger, Caroline He, Morgane Audouard, Arina Nikitina, Stella M K Glasauer, Dasol Han, Barbara Mejía-Cupajita, Juliana Acosta-Uribe, Nelson David Villalba-Moreno, Jessica Lisa Littau , Megan Elcheikhali, Erica Keane Rivera, Daniel Carneiro Carrettiero, Carlos Andrés Villegas-Lanau, Diego Sepulveda-Falla, Francisco Lopera, Kenneth S Kosik
Abstract
Highly penetrant autosomal dominant Alzheimer’s disease (ADAD) comprises a distinct disease entity as compared to the far more prevalent form of AD in which common variants collectively contribute to risk. The downstream pathways that distinguish these AD forms in specific cell types have not been deeply explored. We compared single-nucleus transcriptomes among a set of 27 cases divided among PSEN1-E280A ADAD carriers, sporadic AD, and controls. Autophagy genes and chaperones clearly defined the PSEN1-E280A cases compared to sporadic AD. Spatial transcriptomics validated the activation of chaperone-mediated autophagy genes in PSEN1-E280A. The PSEN1-E280A case in which much of the brain was spared neurofibrillary pathology and harbored a homozygous APOE3-Christchurch variant revealed possible explanations for protection from AD pathology including overexpression of LRP1 in astrocytes, increased expression of FKBP1B, and decreased PSEN1 expression in neurons. The unique cellular responses in ADAD and sporadic AD require consideration when designing clinical trials.

Alzheimer’s & Dementia
Expansion of highly interferon-responsive T cells in early-onset Alzheimer’s disease
Daniel W. Sirkis, Caroline Warly Solsberg, Taylor P. Johnson, Luke W. Bonham, Alexis P. Oddi, Ethan G. Geier, Bruce L. Miller, Gil D. Rabinovici, Jennifer S. Yokoyama
Abstract
Introduction: Altered immune signatures are emerging as a central theme in neurodegenerative disease, yet little is known about immune responses in early-onset Alzheimer’s disease (EOAD).
Methods: We examined single-cell RNA-sequencing (scRNA-seq) data from peripheral blood mononuclear cells (PBMCs) and droplet digital polymerase chain reaction (ddPCR) data from CD4 T cells from participants with EOAD and clinically normal controls.
Results: We analyzed PBMCs from 16 individuals by scRNA-seq and discovered increased interferon signaling-associated gene (ISAG) expression and striking expansion of antiviral-like ISAGhi T cells in EOAD. Isolating CD4 T cells from 19 individuals, including four cases analyzed by scRNA-seq, we confirmed increased expression of ISAGhi marker genes. Publicly available cerebrospinal fluid leukocyte scRNA-seq data from late-onset mild cognitive impairment and AD also revealed increased expression of interferon-response genes.
Discussion: Antiviral-like ISAGhi T cells are expanded in EOAD. Additional research into these cells and the role of heightened peripheral IFN signaling in neurodegeneration is warranted.
Highlights: Interferon-responsive T cells expanded in early-onset Alzheimer’s disease (AD). Increased interferon-associated gene expression present in early- and late-onset AD. Peripheral immune changes in T and NK cells driven by females with early-onset AD.
Keywords: Alzheimer’s disease; CD4 T cells; T cells; cerebrospinal fluid; droplet digital PCR; early‐onset Alzheimer’s disease; interferon; interferon signaling‐associated gene; peripheral blood mononuclear cells; single‐cell RNA‐sequencing; tauopathy.
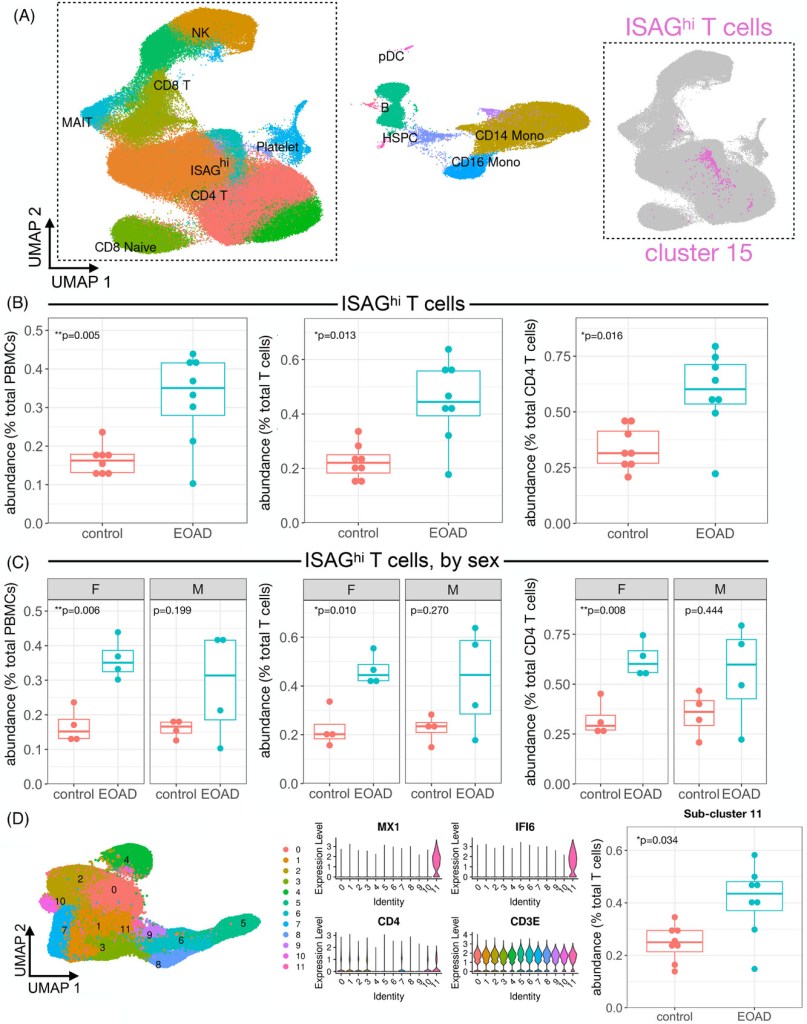
bioRxiv
The wake- and sleep-modulating neurons of the lateral hypothalamic area demonstrate a differential pattern of degeneration in Alzheimer’s disease
Abhijit Satpati, Felipe L. Pereira, Alexander V. Soloviev, Mihovil Mladinov, Eva Larsen, Song Hua Li, Chia-Ling Tu, Renata E. P. Leite, Claudia K. Suemoto, Roberta D. Rodriguez, Vitor R. Paes, Christine Walsh, Salvatore Spina, William W. Seeley, Carlos A. Pasqualucci, Wilson Jacob Filho, Wenhan Chang, Thomas C. Neylan, Lea T. Grinberg
Abstract
Background: Sleep-wake dysfunction is an early and common event in Alzheimer’s disease (AD). The lateral hypothalamic area (LHA) regulates the sleep and wake cycle through wake-promoting orexinergic neurons (OrxN) and sleep-promoting melanin-concentrating hormone or MCHergic neurons (MCHN). These neurons share close anatomical proximity with functional reciprocity. This study investigated LHA OrxN and MCHN loss patterns in AD individuals. Understanding the degeneration pattern of these neurons will be instrumental in designing potential therapeutics to slow down the disease progression and remediate the sleep-wake dysfunction in AD.
Methods: Postmortem human brain tissue from donors with AD (across progressive stages) and controls were examined using unbiased stereology. Formalin-fixed, celloidin-embedded hypothalamic sections were stained with Orx-A/MCH, p-tau (CP13), and counterstained with gallocyanin. Orx or MCH-positive neurons with or without CP13 inclusions and gallocyanin-stained neurons were considered for stereology counting. Additionally, we extracted RNA from the LHA using conventional techniques. We used customized Neuropathology and Glia nCounter (Nanostring) panels to study gene expression. Wald statistical test was used to compare the groups, and the genes were considered differentially expressed when the p-value was <.05.
Results: We observed a progressive decline in OrxN alongside a relative preservation of MCHN. OrxN decreased by 58% (p=0.03) by Braak stages (BB) 1-2 and further declined to 81% (p=0.03) by BB 5-6. Conversely, MCHN demonstrated a non-statistical significant decline (27%, p=0.1088) by BB 6. We observed a progressive increase in differentially expressed genes (DEGs), starting with glial profile changes in BB2. While OrxN loss was observed, Orx-related genes showed upregulation in BB 3-4 compared to BB 0-1. GO and KEGG terms related to neuroinflammatory pathways were mainly enriched.
Conclusions: To date, OrxN loss in the LHA represents the first neuronal population to die preceding the loss of LC neurons. Conversely, MCHN shows resilience to AD p-tau accumulation across Braak stages. The initial loss of OrxN correlates with specific neuroinflammation, glial profile changes, and an overexpression of HCRT, possibly due to hyperexcitation following compensation mechanisms. Interventions preventing OrxN loss and inhibiting p-tau accumulation in the LHA could prevent neuronal loss in AD and, perhaps, the progression of the disease.
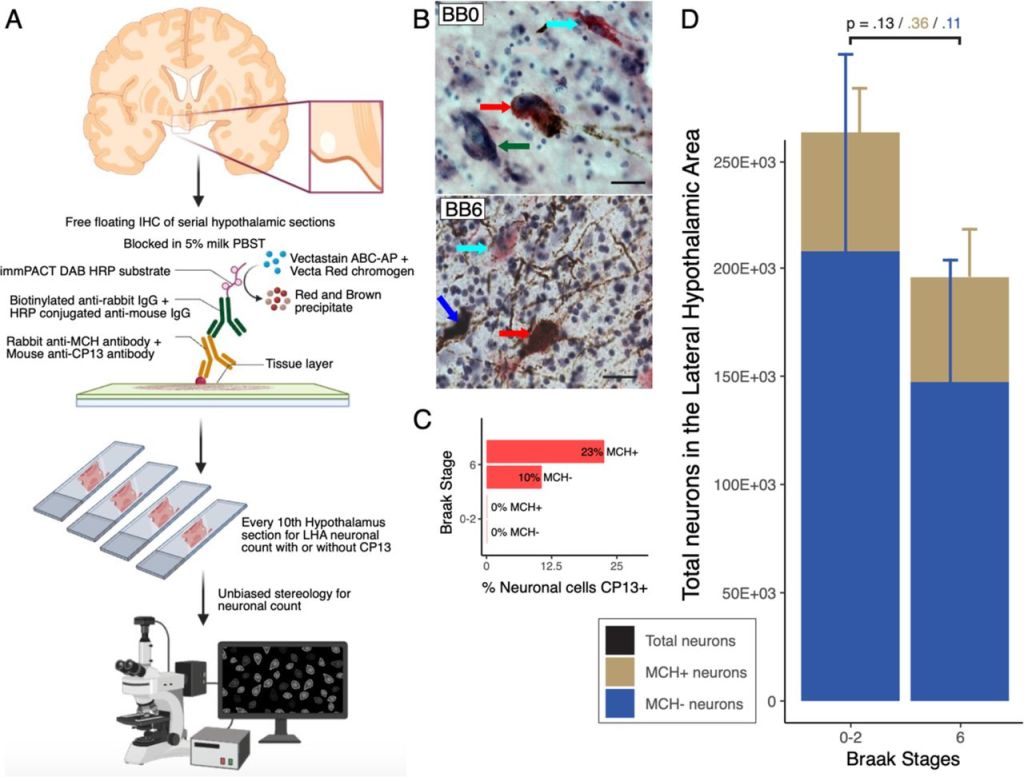
medRxiv
Cell autonomous microglia defects in a stem cell model of frontotemporal dementia
Abhirami K. Iyer, Lisa Vermunt, Farzaneh S. Mirfakhar, Miguel Minaya, Mariana Acquarone, Rama Krishna Koppisetti, Arun Renganathan, Shih-Feng You, Emma P. Danhash, Anthony Verbeck, Grant Galasso, Scott M. Lee, Jacob Marsh, Alissa L. Nana, Salvatore Spina, William W. Seeley, Lea T. Grinberg, Sally Temple, Charlotte E. Teunissen, Chihiro Sato, Celeste M. Karch
Abstract
Neuronal dysfunction has been extensively studied as a central feature of neurodegenerative tauopathies. However, across neurodegenerative diseases, there is strong evidence for active involvement of immune cells like microglia in driving disease pathophysiology. Here, we demonstrate that tau mRNA and protein are expressed in microglia in human brains and in human induced pluripotent stem cell (iPSC)-derived microglia like cells (iMGLs). Using iMGLs harboring the MAPT IVS10+16 mutation and isogenic controls, we demonstrate that a tau mutation is sufficient to alter microglial transcriptional states. We discovered that MAPT IVS10+16 microglia exhibit cytoskeletal abnormalities, stalled phagocytosis, disrupted TREM2/TYROBP networks, and altered metabolism. Additionally, we found that secretory factors from MAPT IVS10+16 iMGLs impact neuronal health, reducing synaptic density in neurons. Key features observed in vitro were recapitulated in human brain tissue and cerebrospinal fluid from MAPT mutations carriers. Together, our findings that MAPT IVS10+16 drives cell-intrinsic dysfunction in microglia that impacts neuronal health has major implications for development of therapeutic strategies.
Journal of Neuroscience
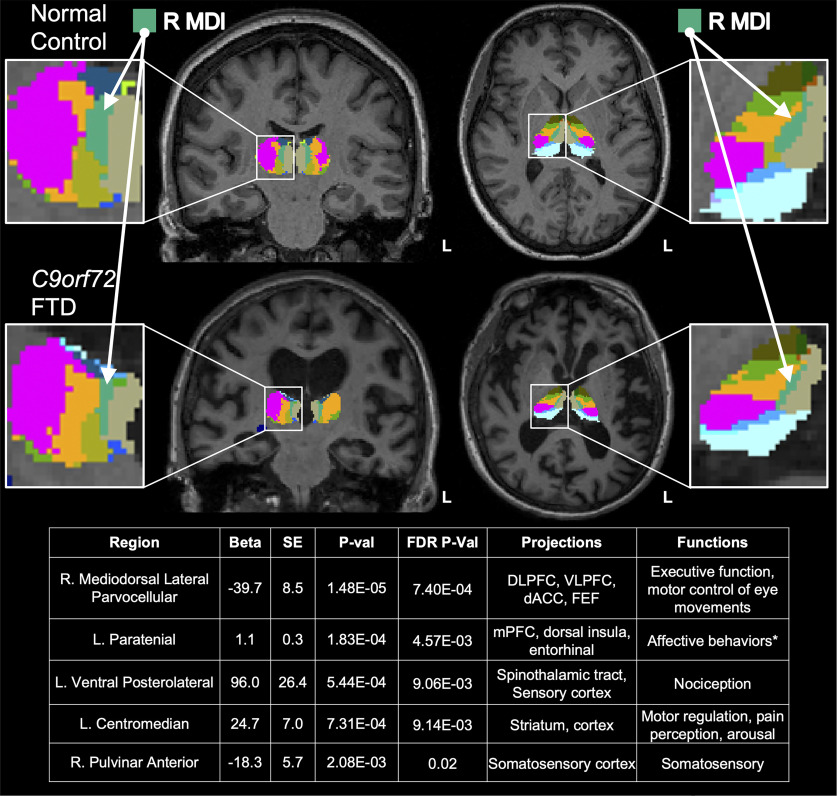
Radiogenomics of C9orf72 Expansion Carriers Reveals Global Transposable Element Derepression and Enables Prediction of Thalamic Atrophy and Clinical Impairment
Luke W. Bonham, Ethan G. Geier, Daniel W. Sirkis, Josiah K. Leong, Eliana Marisa Ramos, Qing Wang, Anna Karydas, Suzee E. Lee, Virginia E. Sturm, Russell P. Sawyer, Adit Friedberg, Justin K. Ichida, Aaron D. Gitler, Leo Sugrue, Michael Cordingley, Walter Bee, Eckard Weber, Joel H. Kramer, Katherine P. Rankin, Howard J. Rosen, Adam L. Boxer, William W. Seeley, John Ravits, Bruce L. Miller and Jennifer S. Yokoyama
Abstract
Hexanucleotide repeat expansion (HRE) within C9orf72 is the most common genetic cause of frontotemporal dementia (FTD). Thalamic atrophy occurs in both sporadic and familial FTD but is thought to distinctly affect HRE carriers. Separately, emerging evidence suggests widespread derepression of transposable elements (TEs) in the brain in several neurodegenerative diseases, including C9orf72 HRE-mediated FTD (C9-FTD). Whether TE activation can be measured in peripheral blood and how the reduction in peripheral C9orf72 expression observed in HRE carriers relates to atrophy and clinical impairment remain unknown. We used FreeSurfer software to assess the effects of C9orf72 HRE and clinical diagnosis (n = 78 individuals, male and female) on atrophy of thalamic nuclei. We also generated a novel, human, whole-blood RNA-sequencing dataset to determine the relationships among peripheral C9orf72 expression, TE activation, thalamic atrophy, and clinical severity (n = 114 individuals, male and female). We confirmed global thalamic atrophy and reduced C9orf72 expression in HRE carriers. Moreover, we identified disproportionate atrophy of the right mediodorsal lateral nucleus in HRE carriers and showed that C9orf72 expression associated with clinical severity, independent of thalamic atrophy. Strikingly, we found global peripheral activation of TEs, including the human endogenous LINE-1 element L1HS L1HS levels were associated with atrophy of multiple pulvinar nuclei, a thalamic region implicated in C9-FTD. Integration of peripheral transcriptomic and neuroimaging data from human HRE carriers revealed atrophy of specific thalamic nuclei, demonstrated that C9orf72 levels relate to clinical severity, and identified marked derepression of TEs, including L1HS, which predicted atrophy of FTD-relevant thalamic nuclei.
SIGNIFICANCE STATEMENT Pathogenic repeat expansion in C9orf72 is the most frequent genetic cause of FTD and amyotrophic lateral sclerosis (ALS; C9-FTD/ALS). The clinical, neuroimaging, and pathologic features of C9-FTD/ALS are well characterized, whereas the intersections of transcriptomic dysregulation and brain structure remain largely unexplored. Herein, we used a novel radiogenomic approach to examine the relationship between peripheral blood transcriptomics and thalamic atrophy, a neuroimaging feature disproportionately impacted in C9-FTD/ALS. We confirmed reduction of C9orf72 in blood and found broad dysregulation of transposable elements-genetic elements typically repressed in the human genome-in symptomatic C9orf72 expansion carriers, which associated with atrophy of thalamic nuclei relevant to FTD. C9orf72 expression was also associated with clinical severity, suggesting that peripheral C9orf72 levels capture disease-relevant information.
July 18, 2023
Genome Medicine
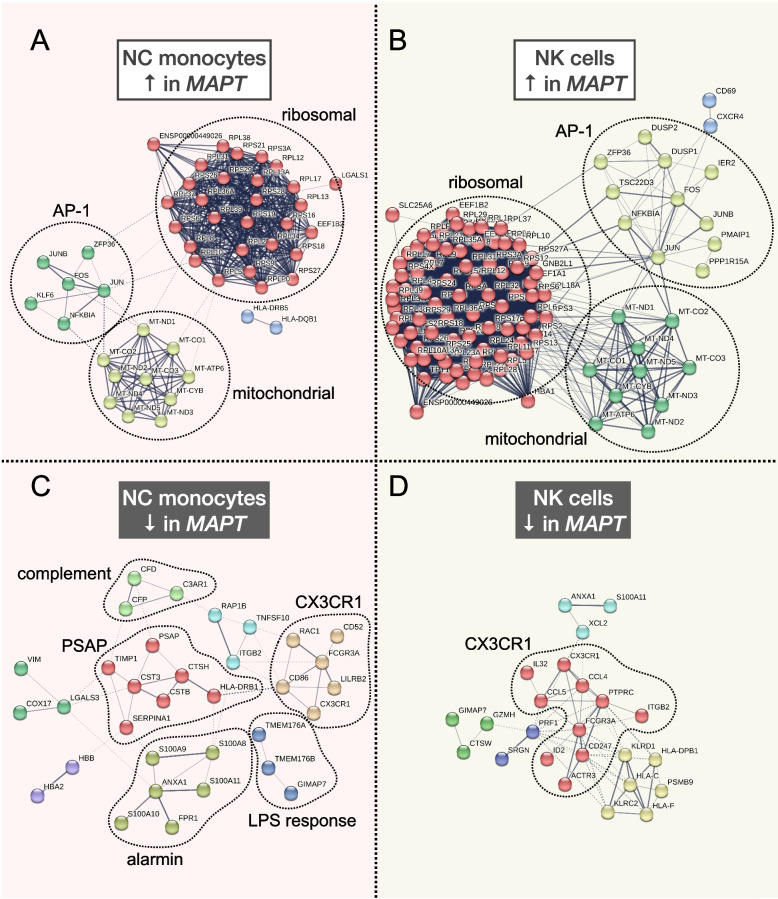
Single-cell RNA-seq reveals alterations in peripheral CX3CR1 and nonclassical monocytes in familial tauopathy
Daniel W. Sirkis, Caroline Warly Solsberg, Taylor P. Johnson, Luke W. Bonham, Virginia E. Sturm, Suzee E. Lee, Katherine P. Rankin, Howard J. Rosen, Adam L. Boxer, William W. Seeley, Bruce L. Miller, Ethan G. Geier & Jennifer S. Yokoyama
Abstract
Background: Emerging evidence from mouse models is beginning to elucidate the brain’s immune response to tau pathology, but little is known about the nature of this response in humans. In addition, it remains unclear to what extent tau pathology and the local inflammatory response within the brain influence the broader immune system.
Methods: To address these questions, we performed single-cell RNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) from carriers of pathogenic variants in MAPT, the gene encoding tau (n = 8), and healthy non-carrier controls (n = 8). Primary findings from our scRNA-seq analyses were confirmed and extended via flow cytometry, droplet digital (dd)PCR, and secondary analyses of publicly available transcriptomics datasets.
Results: Analysis of ~ 181,000 individual PBMC transcriptomes demonstrated striking differential expression in monocytes and natural killer (NK) cells in MAPT pathogenic variant carriers. In particular, we observed a marked reduction in the expression of CX3CR1-the gene encoding the fractalkine receptor that is known to modulate tau pathology in mouse models-in monocytes and NK cells. We also observed a significant reduction in the abundance of nonclassical monocytes and dysregulated expression of nonclassical monocyte marker genes, including FCGR3A. Finally, we identified reductions in TMEM176A and TMEM176B, genes thought to be involved in the inflammatory response in human microglia but with unclear function in peripheral monocytes. We confirmed the reduction in nonclassical monocytes by flow cytometry and the differential expression of select biologically relevant genes dysregulated in our scRNA-seq data using ddPCR.
Conclusions: Our results suggest that human peripheral immune cell expression and abundance are modulated by tau-associated pathophysiologic changes. CX3CR1 and nonclassical monocytes in particular will be a focus of future work exploring the role of these peripheral signals in additional tau-associated neurodegenerative diseases.
Keywords: CX3CR1; Dementia; MAPT; Microglia; Neurodegeneration; Nonclassical monocytes; PBMCs; Single-cell RNA-seq; Tau; Tauopathy.
Stem Cell Reviews and Reports
Postmortem Human Dura Mater Cells Exhibit Phenotypic, Transcriptomic and Genetic Abnormalities that Impact their Use for Disease Modeling
Andrea R Argouarch, Nina Schultz, Andrew C Yang, Yeongjun Jang, Kristle Garcia, Celica G Cosme, Christian I Corrales, Alissa L Nana, Anna M Karydas, Salvatore Spina, Lea T Grinberg, Bruce Miller, Tony Wyss-Coray, Alexej Abyzov, Hani Goodarzi, William W Seeley, Aimee W Kao
Abstract
Patient-derived cells hold great promise for precision medicine approaches in human health. Human dermal fibroblasts have been a major source of cells for reprogramming and differentiating into specific cell types for disease modeling. Postmortem human dura mater has been suggested as a primary source of fibroblasts for in vitro modeling of neurodegenerative diseases. Although fibroblast-like cells from human and mouse dura mater have been previously described, their utility for reprogramming and direct differentiation protocols has not been fully established. In this study, cells derived from postmortem dura mater are directly compared to those from dermal biopsies of living subjects. In two instances, we have isolated and compared dermal and dural cell lines from the same subject. Notably, striking differences were observed between cells of dermal and dural origin. Compared to dermal fibroblasts, postmortem dura mater-derived cells demonstrated different morphology, slower growth rates, and a higher rate of karyotype abnormality. Dura mater-derived cells also failed to express fibroblast protein markers. When dermal fibroblasts and dura mater-derived cells from the same subject were compared, they exhibited highly divergent gene expression profiles that suggest dura mater cells originated from a mixed mural lineage. Given their postmortem origin, somatic mutation signatures of dura mater-derived cells were assessed and suggest defective DNA damage repair. This study argues for rigorous karyotyping of postmortem derived cell lines and highlights limitations of postmortem human dura mater-derived cells for modeling normal biology or disease-associated pathobiology.

